Validation fonctionnelle de QTLs de recombinaison chez S.cerevisiae au sein de l’équipe BASE - Biologie de l’Adaptation et Systèmes en Évolution.
Recrutement en CDD pour 16 mois, niveau Ingénieur.e dans le cadre du projet ANR EVOLREC.
Prise de fonction possible à partir du 1er Janvier 2023
Contexte scientifique
La recombinaison méiotique via les crossing-overs (CO) est un mécanisme clé pour l’évolution des génomes et la dynamique de l’adaptation. Cependant, le nombre et la position des CO sont étroitement régulés mais les pressions de sélection qui ont abouti à cette régulation sont encore largement hypothétiques. En fait la question est complexe car les COs peuvent par exemple accélérer l’adaptation en exploitant mieux la diversité génétique, et inversement, les processus évolutifs de l’adaptation peuvent indirectement sélectionner des modifications du taux de COs.
Le projet pose donc les questions scientifiques suivantes :
- Est-ce que le taux de recombinaison peut évoluer en réponse à une sélection directionnelle ?
- Quels sont les gènes ou autres éléments génétiques qui contrôlent le nombre et la position des COs en cis (à proximité sur le chromosome) et en trans (par ex. sur un autre chromosome) ?
- Quelles sont les inter-relations entre taux de recombinaison et adaptation au stress
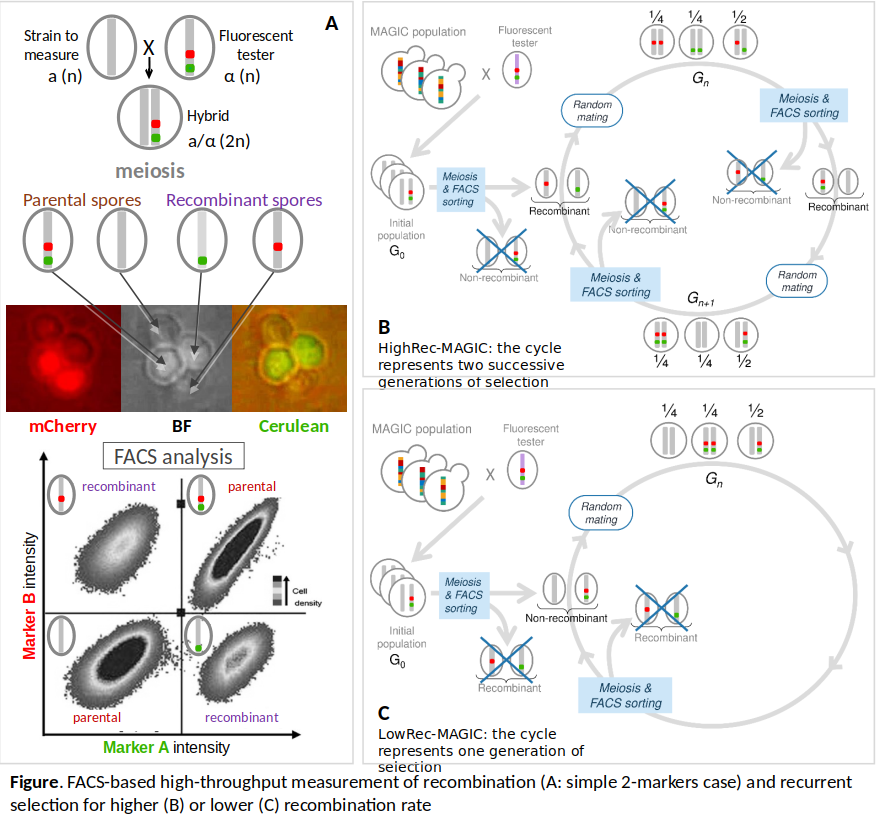
Pour répondre à ces questions expérimentalement chez la levure S. cerevisiae, nous avons développé une méthode pour mesurer à haut-débit le taux de recombinaison par cytométrie en flux avec des marqueurs fluorescents. [1, 2].
Nous avons ensuite réalisé 10 générations de sélection récurrente divergente sur le taux de recombinaison en mode de reproduction sexuée (cf figure ci-dessous) à partir d’une population de levure à forte diversité génétique.
Les populations ainsi sélectionnées ont été séquencées en pool et nous avons identifié des QTLs de recombinaison (agissant en cis et en trans) sur la base des pics de fréquence allélique apparaissant dans les populations sélectionnées.
Approches expérimentales et objectif du poste
A partir des QTLs obtenus précédemment, nous avons identifié des gènes candidats.
Maintenant, avec l’aide de la personne qui sera recrutée, nous allons valider la fonction biologique de ces gènes en étudiant leur effet sur la fréquence de recombinaison. Pour cela, nous allons utiliser des approches de génomique fonctionnelle impliquant de la transformation génétique pour remplacer ou surexprimer des allèles. Par exemple nous allons utiliser la recombinaison homologue ou CRISPR-Cas9 pour remplacer l’allèle d’un gène candidat dans la souche parentale à faible recombinaison par l’allèle de la souche à forte recombinaison, et réciproquement. Nous mesurerons alors les changements d’expression géniques et de phénotype. Nous utiliserons aussi une banque de surexpression déjà présente au laboratoire. Les phénotypes de recombinaison seront mesurés à haut-débit dans plusieurs régions génomiques grâce à la méthode que nous avons déjà mise au point, par cytométrie de flux.
Toutes les ressources - matériel biologique, outils et méthodes - nécessaires au projet ont déjà été produites ou utilisées au laboratoire, et nous disposons d’un grand laboratoire bien équipé pour la biologie moléculaire et la microbiologie.
Environnement de travail
Notre laboratoire est situé sur le campus Paris-Saclay qui est un important regroupement d’instituts de recherche et d’enseignement supérieur proche du centre CNRS de Gif-sur-Yvette, de l’Université Paris-Saclay, et à portée de Paris en RER.
Mots-clés
biologie moléculaire, transformation génétique, recombinaison, QTLs, S. cerevisiae
Compétences requises
Mastère ou diplôme d’ingénieur en biologie moléculaire ou microbiologie. Expérience du travail de paillasse nécessaire. Une expérience en validation fonctionnelle ou sur la levure serait un plus. Anglais lu et parlé.
Contacts
Les candidatures doivent être envoyées par e-mail à: Matthieu Falque