
Des chercheurs de GQE-Le Moulon, du LEPSE, de l’Université d’Olso et de l’Université d’Amsterdam, avec l’aide de la plateforme POPS, ont mis en évidence les réseaux de gènes qui sous-tendent la spécificité tissulaire dans deux types de feuilles de maïs. Ces travaux, publiés dans la revue Frontiers in Genetics - Computational Genomics, représentent une avancée importante en biologie des systèmes car elles combinent des données de génomique, d’épigénomique et de transcriptomique pour faire le lien entre les gènes, leurs régions régulatrices et les protéines qui régulent leur expression.
Le développement des organismes multicellulaires requiert l’expression coordonnée de centaines de gènes selon une spatio-temporalité précise. L’expression de ces gènes est régulée, entre autres, par des régions cis-régulatrices dites « distales » car elles sont localisées à distance des gènes qu’elles ciblent. Des protéines particulières – les facteurs de transcription – se fixent sur ces régions, ce qui cause une interaction avec le promoteur des gènes cibles via des boucles de repliement qui peuvent atteindre plusieurs mégabases. Des analyses récentes combinant des informations acquises à l’échelle du génome sur la méthylation de l’ADN, sur l’accessibilité de la chromatine et sur les marques d’histones ont conduit à la caractérisation de milliers de régions cis-régulatrices distales potentielles chez les plantes.
Chez le maïs (Zea mays ssp. mays), la distance entre un gène et sa région cis-régulatrice distale est en moyenne de 60 kb. Les analyses de repliement tridimensionnel de la chromatine ont montré que plus d’un quart de ces régions ne ciblaient pas le gène le plus proche, et qu’un tiers semblaient réguler plusieurs gènes. Ces caractéristiques rendent très complexe la détection des couples gène/région régulatrice pour l’ensemble du génome, et les approches de biologie moléculaire (comme les systèmes rapporteurs ou CRISPR-Cas9) restent trop coûteuses pour l’analyse conjointe de l’ensemble des gènes.
Dans une étude parue dans Frontiers in Genetics - Computational Genomics, Clémentine Vitte et ses collègues de GQE-Le Moulon, du LEPSE (Montpellier), de l’Université d’Oslo et de l’Université d’Amsterdam ont combiné des données de génomique, d’épigénomique et de transcriptomique afin de reconstruire les liens entre les gènes, leurs régions cis-régulatrices distales et les facteurs de transcription qui se fixent sur celles-ci. Ils ont ainsi mis en évidence des réseaux de gènes exprimés de façon tissu-spécifique dans deux types de feuilles de maïs. L’annotation fonctionnelle de ces réseaux a révélé que dans les jeunes feuilles, les gènes en cause sont impliqués dans la réponse aux hormones, dans la biogenèse des macromolécules et dans l’assemblage de complexes protéiques, alors que dans les spathes (feuilles transformées) qui entourent l’épi, les gènes identifiés jouent un rôle dans la modification de la paroi cellulaire et dans la réponse aux stress abiotiques. La caractérisation de ces réseaux en termes de fonctions biologiques est une avancée pour la génétique et la physiologie du maïs.
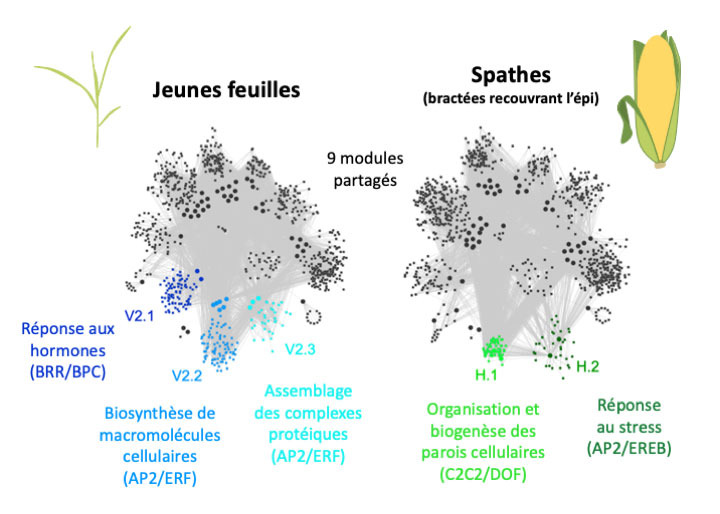
La comparaison des réseaux de régulation des gènes entre les jeunes feuilles (à gauche) et spathes entourant l’épi (à droite) a permis de mettre en évidence neuf modules communs (en noir) et cinq modules spécifiques (en couleurs). Les modules spécifiques des jeunes feuilles (en bleu) sont articulés autour de trois fonctions clés : réponse aux hormones, biosynthèse des macromolécules cellulaires et assemblage des complexes protéiques. Les spathes, elles, sont caractérisées par deux modules (en vert) dont les fonctions clés sont l’organisation et la biogenèse des parois cellulaires et la réponse au stress. Ces fonctions sont en adéquation avec la biologie des deux types de feuilles. Les familles de facteurs de transcription impliquées dans la mise en place de ces réseaux sont indiquées entre parenthèses.
En outre, la preuve de concept de cette méthodologie, qui peut s’appliquer à d’autres tissus, conditions et espèces de plantes, ouvre des perspectives particulièrement intéressantes pour la compréhension de l’évolution de la régulation de l’expression des génomes chez les plantes à fleurs.
Ces travaux ont pu voir le jour grâce au financement du programme AMAIZING, qui a permis la génération de données transcriptomiques sur de nombreux tissus et le recrutement en post-doctorat de Maud Fagny, spécialiste de biologie des systèmes.
Contact : Clémentine Vitte
Référence :