
Dans le cadre de sa collaboration avec Catherine Juste, chercheuse en microbiologie dans l’unité Micalis (INRAE Jouy-en-Josas), PAPPSO a contribué à deux développements en bioinformatique pour l’étude du protéome du microbiote intestinal humain, l’un publié sous forme d’article dans Journal of Proteome Research et l’autre publié sous la forme d’un paquet R sur le CRAN.
Le microbiote intestinal humain, autrefois appelé flore intestinale, est l’ensemble des micro-organismes présents dans notre tube digestif (bouche, estomac, intestin, selles). Ces micro-organismes constituent un véritable écosystème constitué de plus de 4000 espèces. Le microbiote intestinal d’un seul individu peut contenir plus de 600 de ces espèces représentées par plus de cent mille milliards de bactéries, virus, archées et champignons. Les activités du microbiote intestinal sont essentielles pour la digestion et plus généralement pour la physiologie humaine et son influence sur notre santé est maintenant largement reconnue.
Le rôle du microbiote intestinal dans certaines maladies, telles que les maladies inflammatoires de l’intestin ou les maladies cardiovasculaires, reste encore largement à explorer. À ce titre, l’ADN du microbiote intestinal, ainsi que les protéines qu’il exprime, constituent un énorme réservoir de signatures moléculaires qui pourraient servir de biomarqueurs prédictifs ou de nouvelles cibles thérapeutiques pour dépister ou soigner des pathologies.
La plateforme PAPPSO collabore de longue date avec Catherine Juste, notamment dans le cadre des projets Obomics (metaprogramme INRA MEM) et Proteocardis (ANR) impliquant également les cliniciens de l’AP-HP (Pr. Karine Clément, Hôpital Pitié-Salpêtrière ; Pr Harry Sokol, Hôpital Saint-Antoine), afin de rendre possible l’analyse du métaprotéome (i.e. le protéome collectif du microbiote intestinal humain) dans différentes situations physio-pathologiques.
Figure 1: représentation schématique de la stratégie mise en œuvre par Bassignani et al. (2021) pour interpréter les données de métaprotéomique.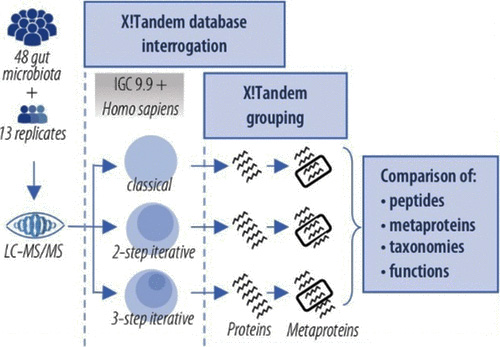
Parallèlement à l’article de Bassignani et al., Aaron Millan-Oropeza et Céline Henry (PAPPSO site de Jouy-en-Josas) publient “metaprotr” sur le CRAN (réseau de serveurs web associé à R). Il s’agit d’un paquet R contenant un ensemble de fonctions permettant l’analyse descriptive des données de métaprotéomique.
L’ensemble de ces développements donnera les moyens d’analyser les données acquises dans le cadre de l’ANR ProteoCardis, un projet ambitieux reposant sur l’analyse de plusieurs centaines d’échantillons de selles humaines afin d’identifier des biomarqueurs de différentes formes de maladies cardiovasculaires.
Référence :
Contacts :
En vignette : Photographie de microbiote purifié par gradient de iodixanol, par Thierry Meylheuc, MIMA2-CIMA platform, INRAE Jouy-en-Josas.